
В мире редких заболеваний есть недуги, о которых не слышало даже большинство врачей, не говоря уже о нас, обычных людях. Болезнь Урбаха-Вите (Урбаха-Витта) - одна из таких загадок.

Это генетическое расстройство, при котором организм буквально «забывает», как правильно обращаться с жирами. Результат - медленное, но неумолимое превращение кожи, слизистых и внутренних органов в плотные, восковые структуры. Почему так происходит, и как живут те, кому поставили такой диагноз?

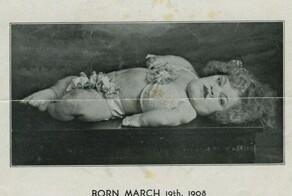
Болезнь названа в честь двух немецких врачей - Эриха Урбаха и Камилло Вите, которые в 1929 году описали первый случай у 11-летней девочки. Её кожа напоминала пергамент, голос был хриплым из-за утолщения голосовых связок, а внутренние органы постепенно приобретали плотность камня. Тогда медики не могли понять причину, но уже знали: это не инфекция и не аллергия. Лишь в 1970-х годах генетики связали болезнь с мутацией гена LIPA, отвечающего за расщепление липидов.

Болезнь Урбаха-Вите проявляется в раннем детстве. В число её основных признаков входят следующие:
- Восковая кожа: эпидермис становится плотным, растягивается, образует глубокие складки, особенно на шее, локтях и коленях.
- Утолщение слизистых: голосовые связки, язык и дыхательные пути теряют эластичность, что приводит к осиплости, затруднённому дыханию.
- Поражение внутренних органов: жировые отложения накапливаются в печени, почках, сердце, нарушая их работу.
- Нейрологические проблемы: судороги, задержка развития из-за повреждения нервной системы.


Пациенты часто становятся пленниками собственного тела: даже мимика и жесты даются с трудом. Всё начинается с мутации в гене LIPA, который кодирует фермент лизосомальную кислую липазу. Этот фермент расщепляет жиры (липиды) в клетках. Если он не работает, липиды накапливаются в лизосомах - «мусорных пакетах» клетки. Со временем это приводит к их разрушению и замене жировыми массами.
Наследуется болезнь по аутосомно-рецессивному типу: ребёнок должен получить дефектный ген от обоих родителей. Шанс такого совпадения составляет 25 %, если оба родителя - носители мутации.

Постановка диагноза осуществляется на основе клинической картины (характерных изменений кожи и слизистых), биопсии и генетического теста, подтверждающего мутации в гене LIPA.
Проблема состоит ещё и в том, что симптомы похожи на другие редкие болезни (например, липоидный протеиноз кожи). Многие пациенты годами блуждают по врачам, прежде чем узнают правду.
К сожалению, болезнь Урбаха-Вите неизлечима. Терапия направлена на облегчение симптомов. Это увлажняющие кремы, местные стероиды, хирургическое удаление избыточных тканей, трахеостомия, противосудорожные препараты.

В США тестируют генную терапию, чтобы «починить» сломанный ген. Пациенты с болезнью Урбаха-Вите сталкиваются не только с физическими муками, но и с социальной стигмой. Их часто принимают за жертв ожогов или неизвестных инфекций. Люди пялятся, шепчутся, тычут пальцами, убегают…

Но самое страшное, что пациенты, страдающие болезнью Урбаха-Вите, не имеют чувства страха из-за отсутствия миндалевидного тела в мозге, которое разрушается вследствие болезни. Они могут выглядеть или казаться абсолютно полноценными и адекватными людьми, пока не встречаются с опасной угрозой, при виде которой обычный человек сбежит.
А такие пациенты её не ощущают и не идентифицируют. Они могут спокойно шагнуть в горящий дом, взять в руки ядовитую змею или сунуть пальцы в розетку. Патологическое бесстрашие сопровождает пациентов до конца их жизни. И никакие факторы – ни приобретённый жизненный опыт, ни здравый смысл, ни советы других не могут на него повлиять.

Но надежда есть. И хотя в медицине зафиксировано и описано всего лишь около 300 случаев этого заболевания, исследования продолжаются. И новейшие исследования показали, что испугать больных возможно, если использовать для этого ингаляцию воздуха с высоким содержанием углекислого газа - около 35 %.

Болезнь Урбаха-Вите напоминает нам о том, как хрупка и сложна наша биология. Каждый ген, каждый фермент – важная составляющая баланса. Изучая такие редкие недуги, учёные приближают день, когда генная терапия сможет исправлять ошибки в ДНК.
Источник:
- Мисс Солнышко – мудрая женщина в теле ребёнка
- Маленький британец уснул и потерял возможность двигаться и говорить
- Зелёные влюблённые: что собой представлял хлороз, который медики называли любовной горячкой?
- Ральф Крунер – человек со слоновьей кожей
- Странная болезнь превратила половину доминиканской семьи в пришельцев
























































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
__________________________________________________________________________________________________
То есть ученые занимаются не поисками возможности излечения, а способом как напугать больного человека?